
NRF2 in COST Action 20121
Our quarterly newsletter attempts to provide our latest news and also aims at becoming a forum for analysis of relevant topics on the field of NRF2 and provide comments to some of the most relevant articles published during the quarter. Previous newsletters can be accessed at:
https://benbedphar.org/our-first-newsletter/
https://benbedphar.org/issue-2-abril-2022/
https://benbedphar.org/issue-3-july-2022/
https://benbedphar.org/issue-4-october-2022/
https://benbedphar.org/issue-5-january-2023/
https://benbedphar.org/issue-6-april-2023/
https://benbedphar.org/issue-7-october-2023/
https://benbedphar.org/issue-8-january-2024/
https://benbedphar.org/issue-9-april-2024/
https://benbedphar.org/issue-10-october-2024/
https://benbedphar.org/issue-11-january-2025/
https://benbedphar.org/issue-12-may-2025/
Antonio Cuadrado
Chair of COST Action 20121, BenBedPhar
Autonomous University of Madrid
Comments from the Working Groups
PIN1-NRF2: A Conditional Partnership
While KEAP1 (Kelch-like ECH-associated protein 1) is recognized as the primary and constitutive repressor of NRF2 (Nuclear factor erythroid-related factor 2) under basal conditions, the functional network of NRF2 is much more complex. Over the past decades, research has demonstrated that NRF2 interacts with a vast network of protein partners, many of which act as conditional regulators, influencing its transcriptional activity, stability, and subcellular localization. Understanding how these conditional protein-protein interactions (PPIs) regulate NRF2 is crucial for comprehending its dual role in health and disease, particularly in the context of its tumor suppressor function in healthy cells versus its oncogenic role in tumor cells. Among these conditional regulators, PIN1 (Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1) has recently emerged as a key modulator of NRF2 function, particularly in cancer.
PIN1 is a peptidyl-prolyl isomerase that catalyzes the cis–trans isomerization of phosphorylated proteins, affecting their stability, activity, and interactions. It recognizes explicitly phosphorylated serine-proline and/or threonine-proline (pS/pT-P) motifs, of which NRF2 has thirteen potential sites that can be isomerized upon phosphorylation by endogenous kinases¹. As a result, the NRF2–PIN1 interaction seems to rely on phosphorylation, making PIN1 a conditional partner of NRF2 that adjusts its activity depending on specific cellular conditions (Fig. 1). The first clear evidence came from studies on triple-negative breast cancer, where activation of H-Ras increased PIN1–NRF2 PPIs through the MAPK cascade1. This stabilized NRF2, promoting its accumulation in the nucleus and encouraging cell proliferation and invasion. Silencing PIN1 reduced NRF2 protein levels and slowed tumor growth1. Follow-up studies showed that phosphorylation of NRF2 at Ser215, Ser408, and Ser577—potentially mediated by ERK and JNK MAPKs—is critical for PIN1 recognition2. Based on the limited literature on PIN1-NRF2 PPIs, it has been proposed that ERK, JNK, and Cdk5 may regulate NRF2 phosphorylation and help assemble a multiprotein complex: Hsp90α-p23-Pin1-Nrf2-importins3 (Fig. 1). This complex could be associated with dynein motors for nuclear transport along microtubules3; however, this idea is only partially supported and still requires experimental validation.
In our recently published structural and biophysical study, direct physical interactions and binding affinities between PIN1 and NRF2-mimicking phosphopeptides were identified, including key domains and residues that govern this interaction4. On NRF2, the Neh7 and Neh3 domains, as well as a region residing between Neh6 and Neh1, are essential. In contrast, on PIN1, residues Ser16, Arg17, Ser18, Tyr23, Ser32, Gln33, and Trp34 within the WW domain form the recognition pocket4 (Fig. 1). Pintide, a peptide targeting the WW domain, effectively disrupts the binding of PIN1 to NRF2. Pharmacological profiling confirmed that small-molecule inhibitors, including juglone, EGCG, and KPT-6566, impair PIN1–NRF2 interactions. Among them, KPT-6566 proved to be the most potent. Mass spectrometry suggested that KPT-6566 covalently modifies PIN1 through conjugate addition rather than disulfide exchange. This indicates that such pharmacophores could serve as starting points for designing specific inhibitors of the PIN1–NRF2 PPI4.
Targeting this interaction may provide a new therapeutic approach to selectively block NRF2-driven cytoprotective signaling in tumors, while leaving its normal protective functions intact. However, more research is necessary to understand the nature and effects of PIN1–NRF2 PPI in healthy tissues and non-cancerous stress conditions.
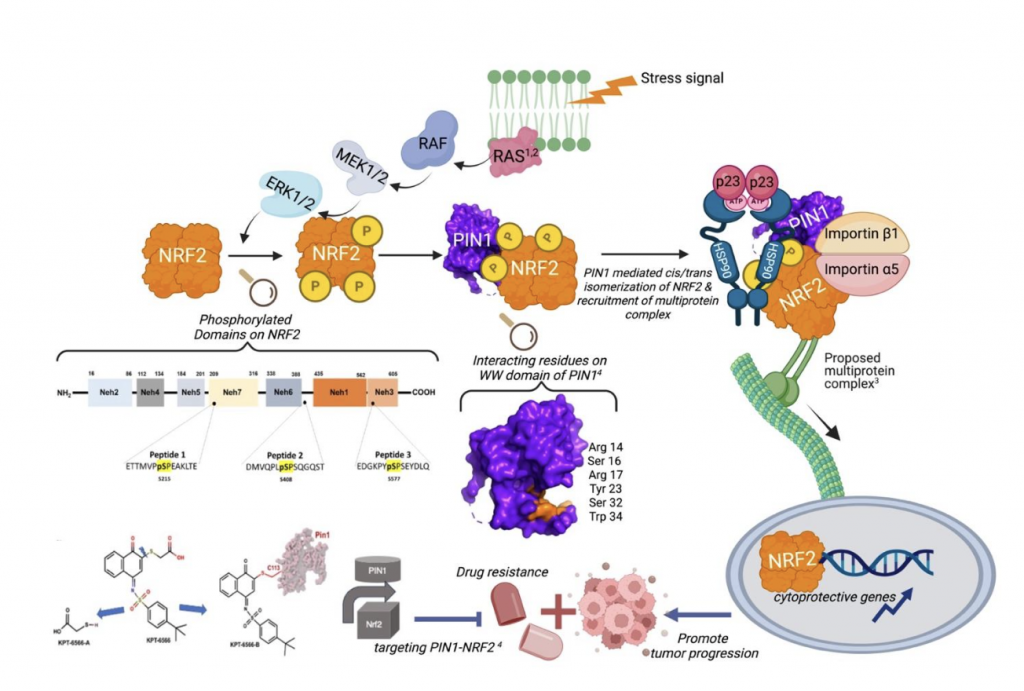
Figure 1. MAPKs activate PIN1-NRF2 PPIs. Phosphorylation of NRF2 by ERK or JNK MAPKs renders it a substrate for PIN1¹,². Recruitment of a multiprotein complex for NRF2 nuclear translocation has been proposed³. Targeting the PIN1–NRF2 PPI may represent a therapeutic approach for stress-associated diseases⁴. Created in BioRender. Tumer, T. (2025) https://BioRender.com/r9bnl65
References:
- Saeidi, S. et al. H-Ras induces NRF2-PIN1 interaction: implications for breast cancer progression. Toxicol. Appl. Pharmacol. 402, 115121 (2020). https://doi.org/10.1016/j.taap.2020.115121.
- Saeidi, S. et al. Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 directly binds and stabilizes NRF2 in breast cancer. FASEB J. 36, e22068 (2022). https://doi.org/10.1096/fj.202101614R.
- Ishii, T., Warabi, E. & Mann, G.E. Stress-activated MAP kinases and cyclin-dependent kinase 5 mediate nuclear translocation of Nrf2 via Hsp90α-Pin1-dynein motor transport machinery. Antioxidants 12, 274 (2023). https://doi.org/10.3390/antiox12020274.
- Ozleyen, A., Duran, G.N., Donmez, S. et al. Identification and inhibition of PIN1-NRF2 protein–protein interactions through computational and biophysical approaches. Sci. Rep. 15, 8907 (2025). https://doi.org/10.1038/s41598-025-89342-0
Tugba Boyunegmez Tumer
Canakkale Onsekiz Mart University, Turkiye
WG1, on behalf of authors
KEAP1 inhibition as a contributor to the therapeutic efficacy of KRASG12C-targeting anti-cancer drugs
The G12C mutation in KRAS occurs in 12-14% of non-small cell lung cancers (NSCLCs), 3-4% of colorectal cancers (CRCs), and 2% in other solid tumors [1]. This cysteine substitution has been exploited for the therapeutic development of selective covalent inhibitors of KRAS, leading to the clinical approval of sotorasib and adagrasib for the treatment of patients with KRASG12C-mutant locally advanced or metastatic NSCLC who have received at least one prior systemic therapy and, in combination with panitumumab, for the treatment of adults with KRASG12C-mutant metastatic colorectal cancer, who have received prior chemotherapy. Interestingly, preclinical studies have shown that, in addition to causing cancer cell death as a monotherapy, sotorasib treatment profoundly affects immune cell infiltration, rendering the tumour microenvironment highly sensitive to immunotherapy, but the underlying mechanism is incompletely understood [2].
In a recent study, Baird et al. [3] found that, at physiologically-relevant concentrations, sotorasib and adagrasib (Fig. 1A) also inhibit KEAP1 by binding to its cysteine sensors (Fig. 1B), resulting in upregulation of NRF2-driven transcriptional program. In cancer cells, NRF2 activation activates the NRF2-induced secretory phenotype (NISP) [4], promoting immune surveillance. In immune cells, NRF2 activation promotes anti-cancer immunity by increasing antioxidant defences and repolarizing myeloid cells towards anti-cancer, pro-inflammatory phenotype, whilst decreasing immunosuppressive IL10 and TGFβ signaling. Overall, in addition to inhibiting oncogenic KRAS signaling, due to their electrophilic chemical properties, the KRASG12C inhibitors sotorasib and adagrasib also inhibit the sensor for electrophiles, KEAP1, leading to NRF2 activation, which in turn promotes anti-cancer immunity and thus contributes to the clinical efficacy of these anti-cancer drugs.
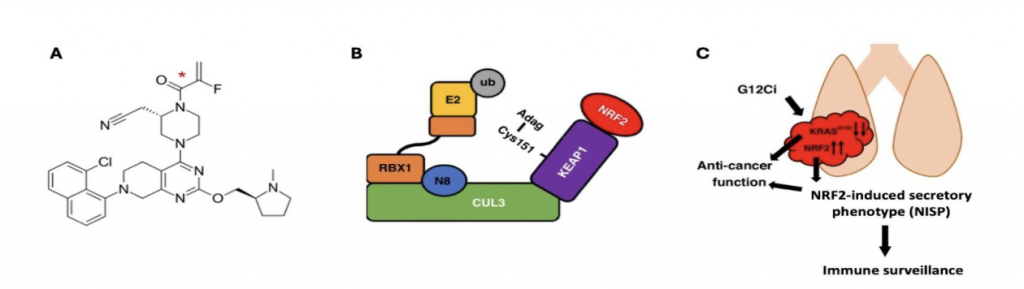
Figure 2. (A) Chemical structure of adagrasib. (B) Adagrasib inhibits KEAP1 by binding to C151, leading to NRF2 activation. (C) NRF2 activation promotes anti-cancer immunity. Adapted from Reference 3.
References:
- AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017; 7: 818-831.
- Cannon et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019; 575: 217-223.
- Baird et al. Systemic activation of NRF2 contributes to the therapeutic efficacy of clinically-approved KRAS-G12C anti-cancer drugs. Br J Cancer. 2025 Sep 1. doi: 10.1038/s41416-025-03162-7.
- Baird et al. A NRF2-induced secretory phenotype activates immune surveillance to remove irreparably damaged cells. Redox Biol. 2023; 66: 102845.
Albena T Dinkova-Kostova
University of Dundee, United Kingdom
WG2 Leaders, on behalf of authors
Successful Short-Term Scientific Mission (STSM) in Finland
From August 25th to 29th, 2025, Christina Morgenstern from the Vienna Airway Lab (Medical University of Vienna) visited the Cellular Neurobiology Lab at the University of Eastern Finland in Kuopio, led by Prof. Katja Kanninen. This visit, part of a Short-Term Scientific Mission (STSM), successfully launched a new collaboration focused on non-invasive biomarker discovery in chronic inflammatory and neurodegenerative diseases. The project explored nasal biofluids – an easily accessible sample type – to identify metabolic biomarkers linked to the NRF2-mediated oxidative stress response, with potential relevance to Chronic Rhinosinusitis (CRS) and Alzheimer’s Disease (AD).
A major outcome was the optimization of a standardized protocol for processing nasal samples using two collection devices – nasosorptions and cytobrushes – enabling detailed metabolomic comparison between samples from a CRS patient and healthy controls. Christina was introduced to advanced computational workflows and bioinformatics tools by bioinformatician Aino-Kaisa Piironen, which led to the detection of several candidate metabolites that may help distinguish disease and healthy states. These results not only provide a foundation for larger studies exploring chronic inflammatory diseases but also demonstrate the potential of nasal fluid as a valuable medium for biomarker discovery.
The mission strengthened interdisciplinary synergy by combining the Vienna Airway Lab’s clinical expertise in respiratory diseases with the Cellular Neurobiology Lab’s specialized knowledge in metabolomics and neurodegeneration. It also supported the career development of early-stage researchers through hands-on training in high-throughput mass spectrometry data analysis and experimental design. The outcomes align closely with the objectives of the BenBedPhar COST Action, providing a strong basis for future collaborative grant applications and scientific publications.

Figure 3. The Cellular Neurobiology Lab with STSM candidate Christina Morgenstern. Front row, from left to right: Aino-Kaia Piironen, Christina Morgenstern, Katja Kanninen
Image Credit: Christina Morgenstern
Christina Morgenstern
Medical University of Vienna, Austria
WG3 Leader
Recent studies have uncovered new therapeutic angles for targeting the NRF2 pathway in cancer
Since the discovery of the association between KEAP1 mutations and the presence of cancer, there has been a revolution in the use of Nrf2 modulators for cancer therapy. This breakthrough has reshaped our understanding of redox biology and cellular defense mechanisms, opening the door to innovative therapeutic strategies. Here, we present new advances in this field, highlighting observations that underscore its potential to transform cancer treatment.
In oncology, the discovery that KRASG12C inhibitors (sotorasib, adagrasib) also induce NRF2 activity highlights a surprising dual mechanism. These drugs covalently bind cysteine residues to inhibit mutant KRAS but simultaneously interact with KEAP1 cysteines, stabilizing NRF2 and triggering its transcriptional program. While chronic NRF2 activation is generally associated with tumor progression and therapy resistance, systemic NRF2 induction by these electrophilic inhibitors appears beneficial. Studies show NRF2 activation not only in tumor cells but also in immune cells within the microenvironment, where it enhances oxidative stress responses and supports anti-cancer immunity. This paradoxical effect may improve treatment outcomes, particularly in patients with KEAP1–NRF2 pathway alterations and has implications for ongoing combination trials.
These findings expand the therapeutic landscape of NRF2 modulation and KRASG12C redefines its role in oncology, suggesting context-dependent benefits. Exploiting this complexity could inform precision strategies cancer therapy.
This fascinating discovery could represent an important step forward in our understanding and development of cancer therapies. However, several concerns arise for researchers investigating the regulation of Nrf2 and its application in different diseases. Genetic activation of Nrf2 has been associated with cancer development suggesting that chronic pharmacological activation of Nrf2 may, under certain conditions, promote tumorigenesis. This remains an unresolved issue, and therefore, these concerns require careful consideration when designing future treatments, both in oncology and in other diseases.
Ioannis Trougakos
The National and Kapodistrian
University of Athens, GR
Santiago Cuevas
BioMedical Research Institute of Murcia, ES
WG4 Co-leaders. Economic Exploitation
BenBedPhar WG5 – Dissemination and Outreach
Throughout the Action, Working Group 5 has played a pivotal role in fostering the exchange of knowledge, tools, and results across BenBedPhar. Its coordinated efforts have greatly enhanced the visibility of the network among diverse stakeholders—including researchers, clinicians, patient associations, the pharmaceutical industry, policy makers, and the general public.
By promoting engagement and collaboration, WG5 has supported the launch of joint initiatives, new research projects, and collaborative journal issues dedicated to knowledge dissemination. These activities have strengthened connections within and beyond the Action, ensuring the long-term sustainability of BenBedPhar as a dynamic and enduring platform for scientific exchange and cooperation.

Figure 5. Working Group 5 (WG5) Aims & Tools for Dissemination
Brigitta Buttari
Istituto Superiore di Sanità, IT
WG5 Leader, on behalf of authors
Most recent (2023–2025) NRF2-modulator clinical trials and near-clinical update.
Direct NRF2 activators
- Omaveloxolone (BIIB141 / SKYCLARYS) – pediatric PK/safety NCT06054893 (2024→) Phase 1: single/multiple-dose PK and safety in ages 2–15 with Friedreich’s ataxia (FA). (ClinicalTrials.gov)
- Omaveloxolone – pediatric efficacy/safety (BRAVE) Biogen announced a Phase 3 pediatric FA study in June 2025 (press + trial listings), expanding evaluation beyond adults. (Trial page not yet assigned an NCT in public sources I could access.) (investors.biogen.com)
- Omaveloxolone – post-marketing safety registry NCT06623890 (2025→) Real-world, multinational observational registry to track long-term safety in FA (PASS). (ClinicalTrials.gov)
- Omaveloxolone – dysphagia in FA (France) NCT07013292 (2025) Interventional study assessing effect on dysphagia in FA. (ClinicalTrials.gov)
- Stabilized sulforaphane (SFX-01) New healthy-volunteer PK/safety study reported in 2024/2025 (ISRCTN96285652; peer-reviewed article). SFX-01 is a clinical sulforaphane formulation that activates NRF2. (isrctn.com)
- Sulforaphane – chronic kidney disease NCT05153174 (ongoing into 2023–2025): dose-finding/PK in CKD patients. (ClinicalTrials.gov)
- Sulforaphane – autism spectrum disorder NCT07047976 (registered 2025): mechanism/efficacy study in ASD; additional pediatric efficacy RCT NCT06594536 (registered 2025). (ClinicalTrials.gov)
NRF2 pathway “opposite” (lowering NRF2) via KEAP1 activation
- VVD-130037 (Vividion/Bayer) – first-in-human KEAP1 activator Phase 1 (started Sept 2023) in advanced solid tumors; intent is to activate KEAP1 to reduce tumor NRF2 activity in KEAP1/NRF2-activated cancers. (Vividion Therapeutics)
Funding Oportunities in the field
- Horizon Europe (EU Missions & Health Cluster) summarized and updated information at the European Institute for Biomedical Imaging Research Website – https://www.eibir.org/open-funding-calls/ several deadlines
- EU4Health Programme (EU4H) – https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/calls-for-proposals?frameworkProgramme=43332642 several deadlines
- European Research Council (ERC) Grants – https://erc.europa.eu/apply-grant several deadlines
- JPND Joint Programme on Neurodegenerative Disease Research – https://neurodegenerationresearch.eu/initiatives/annual-calls-for-proposals/ opening in 2026
- World Cancer Research Fund International (WCRF) – https://www.wcrf.org/research-policy/our-grant-programmes/regular-grant-programme/ several deadlines
- Global Alliance for Chronic Diseases (GACD) – https://www.gacd.org/funding/current-call-for-applications opening in 2026
- la Caixa Foundation: CaixaResearch Health Programme – https://caixaresearch.org/en/caixaresearch-health-call deadline 19th of November
Other funding opportunities available on databases such as:
GrantForward – https://www.grantforward.com/index
Scientific Research – https://www.scientifyresearch.org/grants/
Hot from Pubmed
Aloesin activates Nrf2 signaling to attenuate obesity-associated oxidative stress and hepatic steatosis in high-fat diet-fed rats
Non-alcoholic fatty liver disease (NAFLD) is a prevalent metabolic disorder characterized by hepatic steatosis, oxidative stress, and chronic inflammation. With limited therapeutic options available, there is growing interest in safe bioactive compounds that target underlying mechanisms. Aloesin, a chromone isolated from Aloe vera, possesses potent antioxidants and hypoglycemic properties; however, its protective effect against NAFLD has not been previously examined. This study investigated the hepatoprotective potential of aloesin in rats with high-fat diet (HFD)-induced NAFLD, focusing on the role of Nrf2 signaling. Adult male Wistar rats were divided into seven groups (n = 8/group): control, control + aloesin (200 mg/kg), HFD alone, HFD + aloesin (50, 100, or 200 mg/kg), and HFD + aloesin (200 mg/kg) + brusatol (0.2 mg/kg, i.p.). Treatments were administered twice weekly for 12 weeks. Aloesin dose-dependently improved metabolic and hepatic profiles, reducing body and liver weights, fasting glucose, insulin, HbA1c, HOMA-IR, and serum and hepatic levels of triglycerides, cholesterol, and LDL-c, with 200 mg/kg showing the greatest efficacy. It increased hepatic glucokinase and decreased G6Pase activity. Liver histology revealed restored architecture and reduced inflammation. Serum ALT, AST, and GGT were significantly lowered. Molecular analyses showed increased nuclear Nrf2 and antioxidant markers (GSH, SOD, HO-1), with suppressed NF-κB, TNF-α, IL-6, Bax, and caspase-3, and upregulated Bcl-2. Aloesin also modulated lipid metabolism by decreasing SREBP1 and increasing PPARα expression. These effects were reversed by brusatol, confirming Nrf2 pathway involvement. In conclusion, aloesin confers potent Nrf2-mediated protection against NAFLD, with 200 mg/kg as the optimal therapeutic dose.Access to the original article:https://pubmed.ncbi.nlm.nih.gov/41057370/
Caspase-8 expression and its Src dependent phosphorylation on Tyrosine 380 triggers NRF2 signaling activation in glioblastoma
Caspase-8 expression is upregulated in many tumors where, despite its canonical apoptotic pathway, it sustains cancer progression promoting cell migration, NF-kB activation and inflammation. Here, we provide the first evidence for a novel role of Caspase-8 in promoting the metabolic rewiring of cancer cells. By performing transcriptomic, proteomic and phosphoproteomic analyses on glioblastoma cellular models, we identify Caspase-8 as an unexpected modulator of NRF2. Here we show that Caspase-8 expression and phosphorylation affect NRF2 activity and mitochondrial homeostasis. Mechanistically, we demonstrate that Src-dependent phosphorylation of Caspase-8 on Tyrosine 380 (Y380), frequently reported in cancers including glioblastoma, sustains mTORC1 activation, thus promoting energy metabolism. mTORC1 activity results in p62 phosphorylation allowing its dependent sequestration of KEAP1 protein and constitutive NRF2 signaling activation, as a consequence. Overall, this work depicted a novel unexpected role for Caspase-8 in the modulation of cancer cell metabolism, bridging together Src, mTORC1 and NRF2 signaling.Access to the original article:https://pubmed.ncbi.nlm.nih.gov/41053176/
Bypassing cisplatin resistance in Nrf2 hyperactivated head and neck cancer through effective PI3Kinase targeting
Background: For patients with head and neck squamous cell carcinoma (HNSCC), failure of definitive radiation combined with cisplatin nearly universally results in death. Although hyperactivation of the Nrf2 pathway can drive radiation and cisplatin resistance along with suppressed anti-tumor immunity, treatment-refractory HNSCC tumors may retain sensitivity to targeted agents secondary to synergistic lethality with other oncogenic drivers (e.g., NOTCH1 mutations).
Methods: Using state of the science mechanistic, metabolomic and spatial transcriptomic approaches combined with preclinical models of HNSCC, we tested whether a novel PI3K inhibitor, gedatolisib, can bypass hyperactivation of the Nrf2 pathway.
Results: The PI3K pathway is activated in Nrf2-driven cisplatin-resistant HNSCC and is suitable for blockade, as demonstrated in an in vivo shRNA screen with platinum-based chemotherapy. Gedatolisib effectiveness appears mediated through activation of autophagy, G2/M arrest, senescence and disruption of fatty acid metabolism. Gedatolisib suppresses HNSCC tumor growth in orthotopic and metastatic settings and demonstrates profound anti-tumor activity in humanized murine models of HNSCC, coupled with a reduction in hypoxia-rich regions and reduced infiltration by regulatory T-lymphocytes.
Conclusions: These findings emphasize the critical role of the PI3K-AKT-mTOR pathway in chemo-radiation resistant HNSCC and highlight the therapeutic potential of PI3K inhibitors in a disease that is refractory to all conventional therapeutic approaches.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/41053162/
Advances in mitochondria-nucleus crosstalk in septic cardiomyopathy
Sepsis-induced cardiomyopathy (SICM), a critical contributor to the high mortality rate associated with sepsis, involves complex pathophysiological mechanisms that remain incompletely elucidated. In recent years, dysregulation of bidirectional signaling communication between mitochondria and the nucleus has been recognized as a pivotal factor in the pathogenesis of SICM. The anterograde signaling pathways-including the PGC-1α/NRF1/NRF2 axis, SIRT3-mediated deacetylation, and TFAM-dependent mitochondrial DNA (mtDNA) maintenance-are suppressed by inflammation and metabolic disturbances. This suppression leads to impaired mitochondrial biogenesis and disrupted energy metabolism. Concurrently, within retrograde signaling pathways, molecular mediators such as reactive oxygen species (ROS), mtDNA, and calcium signaling activate pro-inflammatory and apoptotic pathways, notably NF-κB and cGAS-STING. This activation establishes a vicious cycle perpetuating inflammation and cellular damage. Although current targeted interventions aimed at modulating mitochondrial-nuclear crosstalk have demonstrated some efficacy in animal models, their clinical translation faces significant challenges. These include the dynamic nature of the disease, substantial interindividual variability, and difficulties in achieving targeted delivery. This review summarizes the mechanisms of mitochondrial-nuclear bidirectional signaling in SICM and explores potential therapeutic targets, aiming to provide novel insights for SICM treatment strategies.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/41051583/
Turning Off the Ferroptosis Switch: ACAA1-Driven PI3K/AKT/Nrf2 Signaling as a Novel Driver of Endometrial Cancer Progression
Endometrial carcinoma (EC) is a prevalent gynecologic malignancy with rising global incidence. Dysregulated lipid metabolism promotes EC progression through estrogen synthesis, metabolic reprogramming, and tumor microenvironment remodeling. Ferroptosis, an iron-dependent cell death driven by lipid peroxidation, represents a potential therapeutic strategy, yet its resistance mechanisms in EC remain unclear. We identify Acetyl-CoA Acetyltransferase 1 (ACAA1), a key enzyme in mitochondrial fatty acid β-oxidation, as an oncogenic factor in EC. We demonstrate that ACAA1 is significantly upregulated in EC tissues via bioinformatic analysis and clinical samples. Functionally, ACAA1 overexpression enhances tumor cell proliferation, migration, energy metabolism, and lipid droplet synthesis in vitro, while accelerating tumor growth in vivo in xenograft models. Mechanistically, ACAA1 activates the PI3K/AKT pathway, leading to nuclear translocation of the transcription factor Nrf2. This ACAA1/PI3K/AKT/Nrf2 axis suppresses ferroptosis by regulating redox homeostasis and lipid peroxidation, thereby promoting EC progression. Our findings reveal ACAA1 as a novel regulator of ferroptosis resistance and tumorigenesis in EC, highlighting its potential as a promising therapeutic target for EC treatment.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/41046947/
Isoliquiritigenin attenuates cisplatin-induced hearing loss and ototoxicity by activating the Keap1-Nrf2-ARE pathway
Cisplatin-induced hearing loss (CIHL), a major dose-limiting toxicity of cisplatin, is primarily caused by oxidative stress and apoptosis in cochlear hair cells. This study aims to investigate the otoprotective effects of Isoliquiritigenin (ISL, a natural Nrf2 agonist) on CIHL and to elucidate the underlying anti-CIHL mechanism(s) of ISL. Initially, ISL was identified as a natural Nrf2 agonist from a phytochemical library using a luciferase reporter gene system. The otoprotective effects of ISL were then investigated in HEI-OC1 cells, cochlear explants, and in cisplatin-induced ototoxicity murine models. In cisplatin-induced ototoxicity mice, ISL markedly restored full-frequency auditory brainstem response (ABR) thresholds and attenuated cisplatin-induced hair cell loss in the cochlea. In HEI-OC1 cells and cochlear explants, ISL significantly attenuated cisplatin-triggered reactive oxygen species (ROS) overproduction, mitochondrial dysfunction, and hair cell apoptosis. Mechanistically, ISL covalently modify two critical cysteine residues (Cys226 and Cys288) of KEAP1, which subsequently stabilized Nrf2 and upregulated the expression of downstream antioxidant proteins including NAD(P)H quinone oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1) and superoxide Dismutase (SOD). Collectively, our findings clearly demonstrate that ISL significantly attenuates cisplatin-induced hearing loss (CIHL) by activating the Keap1-Nrf2-ARE signaling via covalent modifying two key cysteine residues on KEAP1.
Access to the original article:https://pubmed.ncbi.nlm.nih.gov/41052583/
MOTS-c attenuates mitochondrial dysfunction induces pyroptosis and cartilage degradation in osteoarthritis via an Nrf2-Dependent Mechanism
Osteoarthritis, a common chronic degenerative disease in the field of orthopedics, is caused by the interaction of mechanical stress, traumatic inflammation, and metabolic imbalance, and this interaction progresses over time. MOTS-c, a mitochondria-derived peptide, exerts pivotal roles in regulating metabolism, anti-inflammation, and antioxidant stress responses. However, current research on the role of MOTS-c in osteoarthritis remains scarce, and its specific mechanism of action remains unclear. Therefore, this study aims to further explore the molecular mechanisms by which MOTS-c regulates osteoarthritis. Exogenous supplementation of MOTS-c improves mitochondrial dysfunction, inhibits the activation of inflammatory bodies and rescues chondrocyte pyroptosis, thereby regulating the metabolic balance of extracellular matrix (ECM). Mechanistically, MOTS-c plays a key role in LPS-induced oxidative stress and chondrocyte pyroptosis through the Nrf2/TXNIP/NLRP3 axis. Our research demonstrates that MOTS-c can not only effectively inhibit the expression of inflammatory factors but also promote the expression of major components of the extracellular matrix (ECM) and suppress the production of matrix metalloproteinases. We validated the in vivo efficacy of MOTS-c by establishing a murine osteoarthritis model. Analysis of imaging and histopathological results revealed that MOTS-c can effectively delay the degeneration of articular cartilage and ameliorate the progression of osteoarthritis. Collectively, our findings uncover the intrinsic regulatory mechanism of MOTS-c in chondrocytes and its potential value in the treatment of osteoarthritis.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/41043625/
Pioglitazone improves tamoxifen-induced renal injury in rat model through modulating oxidative stress, inflammation, and apoptosis
One prevalent chemotherapeutic medication used to treat breast cancer is tamoxifen (TAM), and several off-label uses. However, when used as an antineoplastic drug in clinical settings, it causes harmful cellular effects such as renal damage. The mechanisms underlying TAM-induced kidney injury primarily involve oxidative stress and inflammatory pathways. Pioglitazone (PIO), an agonist of peroxisome proliferator-activated receptor-gamma (PPAR-γ), is a medication primarily indicated for type 2 diabetes mellitus. Beyond its hypoglycemic actions, PIO exhibits potent anti-inflammatory and antioxidant effects, which have been documented in various tissues. Hence, this study investigates the potential protective effects of PIO on TAM-induced renal impairment, revealing their possible protective mechanisms. The rats were orally administered doses of PIO (10 mg/kg) and TAM (45 mg/kg) over ten days. Renal histopathological changes, kidney function, and biochemical examination are conducted. TAM raised serum creatinine and urea levels, leading to histopathological changes, renal oxidative stress, and elevated expression of NF-κB p65 and proinflammatory cytokines. Conversely, PIO treatment exerted a protective effect and attenuated TAM-induced nephrotoxicity. Significantly, malondialdehyde (MDA) and proinflammatory markers were reduced while enhancing renal antioxidant activity. Furthermore, renal Nrf2, HO-1, Bcl-2 expression while lowering NF-κB, KIM-1, Bax and caspase-3 levels. In conclusion, PIO demonstrated significant nephroprotective effects against TAM-induced renal damage by inhibiting apoptosis, reducing inflammation, and counteracting oxidative stress.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/41037918/
Joana Miranda
Faculty of Pharmacy, University of Lisbon
Portugal