
NRF2 in COST Action 20121
Our quarterly newsletter attempts to provide our latest news and also aims at becoming a forum for analysis of relevant topics on the field of NRF2, and provide comments to some of the most relevant articles published during the quarter. Previous newsletters can be accessed at:
https://benbedphar.org/our-first-newsletter/
https://benbedphar.org/issue-2-abril-2022/
https://benbedphar.org/issue-3-july-2022/
This October issue links with the end of the first period of our COST Action, and provides a perfect opportunity to make some reflections and to have a critical analysis of our activities. In my opinion, our most important success so far is that, after just one year of activity, we are over 230 participants from 33 countries all over Europe. Among them, 67% are women and almost one third are young researchers. Looking back to the beginning of the Action, I think that the COST tools have significantly help to disseminate important concepts about the mechanistic regulation of NRF2, its participation in chronic diseases, and the multiple possibilities of pharmacological regulation.
Many other achievements were also reported during our last Management Committee meeting that was held in mixed format at the Victor Babes National Institute of Pathology, in Bucharest (Romania) in October 13. These achievements included our previous newsletters, webinars, meetings, etc. and an unexpectedly high number of collaborative reviews and some experimental papers published in our guest edited special issues. All these activities have paved the way to strengthen collaborative activities and the MC members encouraged BenBedPhar participants to look for ways to make use of COST tools to increase collaborative research, including STSMs, publication of joint research articles and preparation of grant application to private and public calls. In this regard, at least three grants have been awarded to collaborative projects among BenBedPhar participants.
At the verge of ending the first grant period, we had our scientific meeting, in October 13-15, at the Victor Babes National Institute of Pathology, in Bucharest, with title “Bench to Bedside for Pharmacological regulation of NRF2 in no communicable diseases”. We gathered over 60 participants with 35 oral communications and a few poster presentations. This time the meeting was focused to enhance interactions among participants with the end goal of establishing collaborative agreements. You can access the programme and the abstract book at https://benbedphar.org/3rd-benbedphar-scientific-meeting/.
We are now facing the second grant period with lots of illusion and big expectations to strengthen the research interactions among the EU and international researchers interested in this transcription factor that some days we hate and most days we love. A day will soon come in which NRF2 research will conclusively be translated to the clinic and provide a therapeutic option for many chronic diseases that so badly impact our wellbeing.
Antonio Cuadrado
Chair of COST Action 20121, BenBedPhar
Autonomous University of Madrid
In Memoriam of Prof. Michael Sporn

On September 29, 2022, the NRF2 field lost one of its giant figures, Dr Michael B. Sporn, an internationally recognized cancer researcher, who passed away at his home in Tunbridge, Vermont (USA) at the age of 89. Dr. Sporn’s scientific life was dedicated to developing new approaches for the prevention and treatment of cancer and chronic disease. He was the founding father of the concept of cancer chemoprevention defined as “the use of pharmacologic or natural agents that inhibit the development of invasive cancer either by blocking the DNA damage that initiates carcinogenesis or by arresting or reversing the progression of premalignant cells in which such damage has already occurred”.
Highlighting the importance of cancer prevention and challenging the existing dogma for cancer treatment based on treating end-stage disease, Dr. Sporn stated in his “Plea for Prevention” in a 1996 issue of Scientific American: “An ‘obsession’ with curing advanced disease has blinded cancer researchers to the promise of prevention… The concept that people with cancer are healthy until a doctor tells them that they’ve got an invasive lesion makes no sense at all.”
Dr Sporn graduated from Harvard and subsequently from the University of Rochester School of Medicine. He then worked at the National Cancer Institute (NCI) where he became Chief of the Lung Cancer Branch and later Chief of the Laboratory of Chemoprevention. During that time, he made ground-breaking discoveries on retinoids and Transforming Growth Factor β (TGFβ). In 1995, he became Professor of Pharmacology and Medicine at Dartmouth Medical School where he pioneered the development of the cyanoenone semi-synthetic triterpenoids as extremely potent anti-inflammatory agents and NRF2 activators. To date, this class of compounds represents the most potent NRF2 activators known, which have shown protective effects in numerous models of human disease. In 2018, Dr Sporn founded the company Triterpenoid Therapeutics, Inc. to develop and commercialize new compounds for cancer prevention and treatment.
For his outstanding scientific achievements, Dr Sporn received several prestigious awards, including the inaugural American Association for Cancer Research (AACR) and the Cancer Research Foundation of America Award for Excellence in Cancer Prevention Research, a Medal of Honor from the American Cancer Society, the Bristol-Myers Squibb Award for Distinguished Achievement in Cancer Research, the Lila Gruber Award for Cancer Research from the American Academy of Dermatology, the Komen Brinker Award for Scientific Distinction. In 2004, he was named by the NCI as its first Eminent Scholar. In 2007, he was a member of the Cancer Advisory Board of the President of the United States.
Dr Sporn was a passionate scientist and an enthusiastic mentor and collaborator. He will be greatly missed by the scientific community for his gigantic intellect, enormous contributions and commitment to science, entrepreneurial spirit, and generosity in sharing his ideas, discoveries, and materials, and in supporting the next generation of cancer researchers.
Albena Dinkova-Kostova
WG2 leader
University of Dundee, UK
Comments from the Working groups
Role of inflammation and redox status on doxorubicin-induced cardiotoxicity in infant and adult CD-1 male mice
Doxorubicin (DOX) is a topoisomerase II inhibitor commonly used in the treatment of several types of cancer. Despite its efficacy, DOX can potentially cause fatal adverse effects, like cardiotoxicity. This work aimed to assess the role of inflammation in DOX-treated infant and adult mice and its possible link to underlying cardiotoxicity. Two groups of CD-1 male mice of different ages (infants or adults) were subjected to biweekly DOX administrations, to reach a cumulative dose of 18.0 mg/kg, which corresponds approximately in humans to 100.6 mg/m2 for infants and 108.9 mg/m2 for adults a clinically relevant dose in humans. The classic plasmatic markers of cardiotoxicity increased, and that damage was confirmed by histopathological findings in both groups, although it was higher in adults. Moreover, in DOX-treated adults, an increase of cardiac fibrosis was observed, which was accompanied by an increase in specific inflammatory parameters, namely, macrophage M1 and nuclear factor kappa B (NF-κB) p65 subunit, with a trend toward increased levels of the tumor necrosis factor receptor 2 (TNFR2). On the other hand, the levels of myeloperoxidase (MPO) and interleukin (IL)-6 significantly decreased in DOX-treated adult animals. In infants, a significant increase in cardiac protein carbonylation and in the levels of nuclear factor erythroid-2 related factor 2 (Nrf2) was observed. In both groups, no differences were found in the levels of tumor necrosis factor (TNF-α), IL-1β, p38 mitogen-activated protein kinase (p38 MAPK) or NF-κB p52 subunit. In conclusion, using a clinically relevant dose of DOX, our study demonstrated that cardiac effects are associated not only with the intensity of the inflammatory response but also with redox response. Adult mice seemed to be more prone to DOX-induced cardiotoxicity by mechanisms related to inflammation, while infant mice seem to be protected from the damage caused by DOX, possibly by activating such antioxidant defenses as Nrf2.
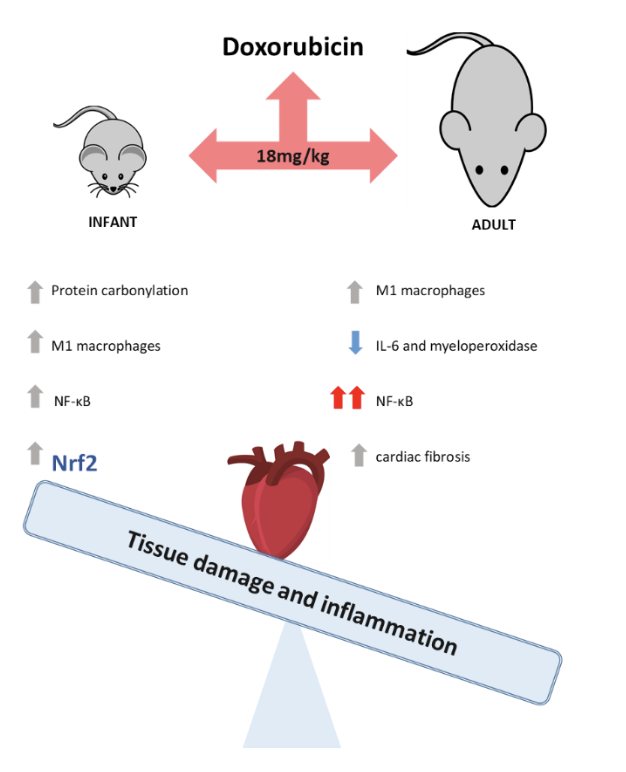
Figure 1. Doxorubicin’s cardiotoxic effects were strongly attenuated in infant mice by Nrf2 activation, whose role on future pharmacological treatment of cancer patients dealing with cardiac adverse effects requires further research.
Vera Marisa Costa
WG1 member, on behalf of authors in
https://doi.org/10.3390/biom11111725
University of Porto, Portugal
Another way to skin the cat: Small molecule inhibitors of the β-TrCP-NRF2 interaction as a strategy for activation of NRF2
Under homeostatic conditions, NRF2 is a short-lived protein that is continuously targeted for ubiquitination and proteasomal degradation. Three known ubiquitin ligase systems mediate the degradation of NRF2: (i) Kelch-like ECH associated protein 1 (KEAP1), a substrate adaptor protein for Cullin3 (Cul3)/Rbx1-based Cullin-RING E3 ubiquitin ligase and a cysteine-based sensor for NRF2 inducers; (ii) the E3 ubiquitin ligase Hrd1, which resides in the endoplasmic reticulum (ER), and degrades NRF2 during ER stress; (iii) β-transducin repeat-containing protein (β-TrCP), a substrate adaptor for Skp1-Cullin1 (Cul1)/Rbx1-based Cullin-RING E3 ubiquitin ligase (Holmström et al. 2016). Notably, degradation of NRF2 by β-TrCP requires the formation of a phosphodegron on NRF2 following phosphorylation catalyzed by glycogen synthase kinase 3 (GSK3) (Rada et al. 2011). Most pharmacological NRF2 activators known to date target KEAP1, by either modifying its cysteine sensors or disrupting its interaction with NRF2. In a new study, Fernández-Ginés et al. adopted an alternative strategy for the development of pharmacological NRF2 activators, namely by targeting the β-TrCP-NRF2 protein-protein interactions (Fernández-Ginés et al. 2022). After in silico screening of ∼1 million compounds, they identified a small molecule, named PHAR (Figure 2), which selectively inhibits the interaction between β-TrCP and the phosphodegron in NRF2. In cells, PHAR was protective against hydrogen peroxide-induced oxidative stress and liposaccharide (LPS)-mediated pro-inflammatory stress. In vivo, intraperitoneal (i.p.) administration of PHAR activated NRF2 in the mouse liver, attenuated the increase of pro-inflammatory cytokines following LPS challenge; this was accompanied by reduced activation of Kupffer cells, the liver resident macrophages. This study supports the value of pharmacological NRF2 activators as protective agents against oxidative stress and inflammation and provides a proof-of-principle that such protection can be achieved without inhibition of KEAP1.
References: Fernández-Ginés R, Encinar JA, Hayes JD, Oliva B, Rodríguez-Franco MI, Rojo AI, Cuadrado A. An inhibitor of interaction between the transcription factor NRF2 and the E3 ubiquitin ligase adapter β-TrCP delivers anti-inflammatory responses in mouse liver. Redox Biol. 2022 Jul 11;55:102396. Holmström KM, Kostov RV, Dinkova-Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016 Dec;1:80-91. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/ β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011 Mar;31(6):1121-33.
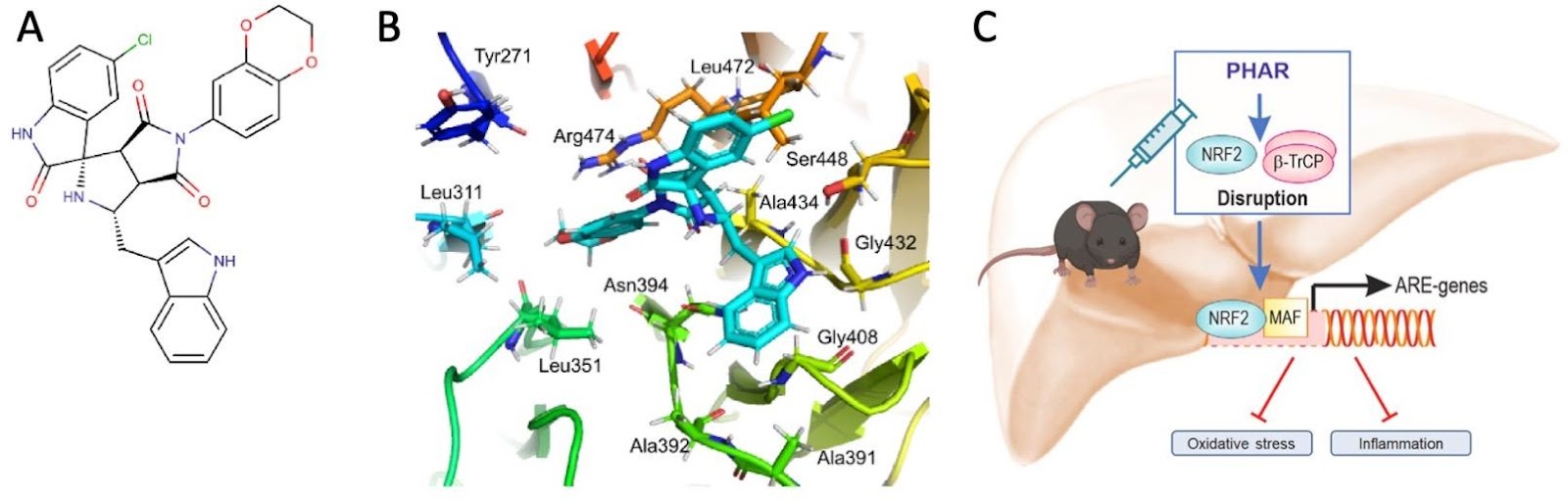
Figure 2: (A) Spatial localisation of the amino acids of β-TrCP that form hydrophobic or electrostatic interactions with PHAR based on molecular docking simulations; (B) PHAR inhibits the interactions between NRF2 and β-TrCP, activates NRF2-mediated transcription in the mouse liver, and suppresses oxidative stress and inflammation. Figure adapted from: Fernández-Ginés et al. 2022.
Albena Dinkova-Kostova
WG2 leader
University of Dundee, UK
A matter of gender: The lack of transcriptionally active Nrf2 triggers colon dysfunction in female mice – The role of estrogens
The proper functioning of the gastrointestinal system relies on an intricate crosstalk between a plethora of cell types and signaling pathways. Recently we identified that the lack of NRF2 transcriptional activity (NRF2 tKO) triggers significant colon microscopical alterations, still they do not affect the general functioning of mice. Therefore, in this study, we aimed to address the gender-dependent impact of NRF2 transcriptional deficiency on colon function and relate them to an established model of inflammatory bowel disease (IBD). In the study we subjected 3- and 6-month-old mice deficient in IL-10 and NRF2 transcriptional activity and wild-type counterparts to tests assessing colon functionality, and histological analyses. To address the role of estrogens, we attempted to rescue the phenotype by the delivery of 17β-estradiol through subcutaneous implants. In females, NRF2 transcriptional abrogation, like IL-10 deficiency, triggers a functional and microscopic phenotype, that resembles IBD. The females are significantly more affected by the dysfunctional phenotype, and the functional impairment decreases with age. We found that NRF2 transcriptional activity influences 17β-estradiol level and the estrogen receptors expression and location. Exogenous delivery of 17β-estradiol normalized colon motility in the NRF2 tKO mice, which is related to enhanced ERβ signaling. Summing up, in this study, we underline that NRF2 transcriptional deficiency or the lack of IL-10 results in pronounced GI functional decline in young females. Mechanistically, we show that the impaired distal colon motility is dependent on ERβ signaling. Targeting estrogen signaling seems a promising therapeutic strategy to counteract colonic dysfunction.
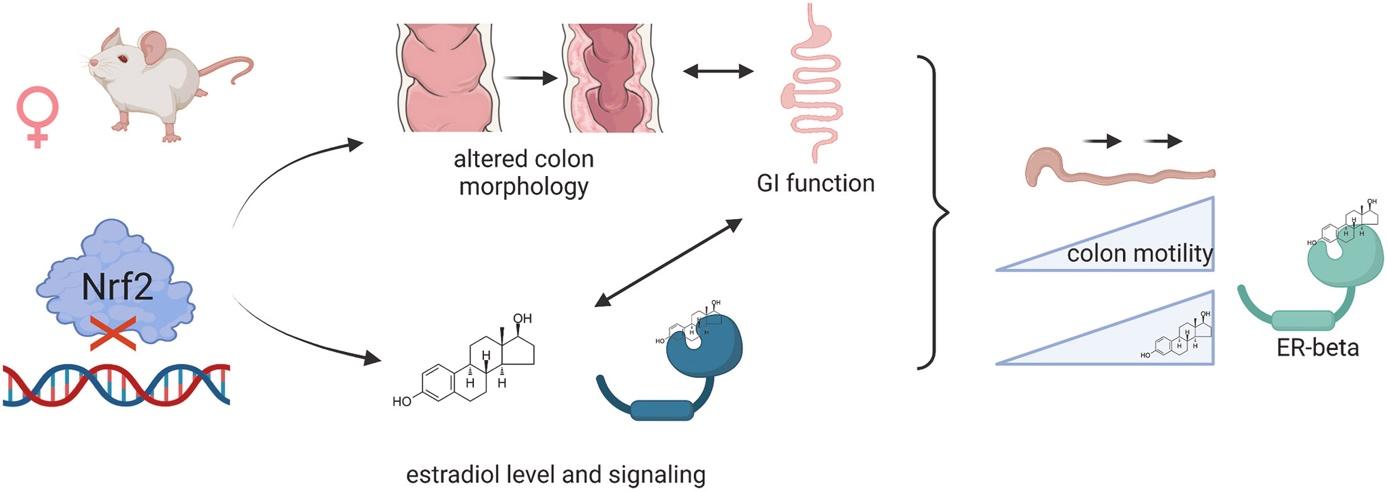
Figure 3. The lack of transcriptionally active Nrf2 triggers colon dysfunction in female mice by influencing levels of 17β-estradiol and the expression of estrogen receptors.
Aleksandra Piechota-Polanczyk
WG3 member on behalf of co-authors in
https://doi.org/10.1016/j.freeradbiomed.2022.09.014
Jagiellonian University, Krakow, Poland
Medical University of Lodz, Poland
The protective role of NRF2 against fibrosis
Fibrosis is characterized by the accumulation of extracellular matrix, often in the context of chronic inflammation, leading to pathological conditions in many organs such the lung, liver and heart. These conditions can be triggered by different stimuli, being oxidative stress, persistent infection, and autoimmune reactions some examples (Cho et al., 2004; Hao et al., 2022; Wynn, 2008). As a key player in the antioxidant defense response, NFR2 plays a major role in preventing the generation of fibrotic tissue. In the myocardium, Nrf2 is responsible for the activation of the glutathione pathway that contributes for the maintenance of the redox balance, preventing increased inflammation, fibroblast activation and myocardial fibrosis. The activation of Nrf2-dependent antioxidant mechanisms and consequent reduction of oxidative stress levels also prevents epithelial-mesenchymal transition, a known characteristic of fibrosis, and acute injury in the lung and reduce insulin resistance and attenuate fibrosis in the liver. Interestingly, Nrf2 role is not limited to antioxidant mechanisms. In fact, Nrf2 was shown to be responsible to inhibit the activation of pathways such as TGF-β/SMADs, which lead to the production and accumulation of collagen. In the lung, Nrf2 is important during the immune response by influencing cytokine production, and, in the liver, Nrf2 knockdown was shown to stimulate extracellular matrix synthesis leading to fibrosis. Not surprisingly, the important role of NRF2 to counter fibrosis has promoted several research groups to explore the potential of targeting NRF2 pathway to treat fibrosis.
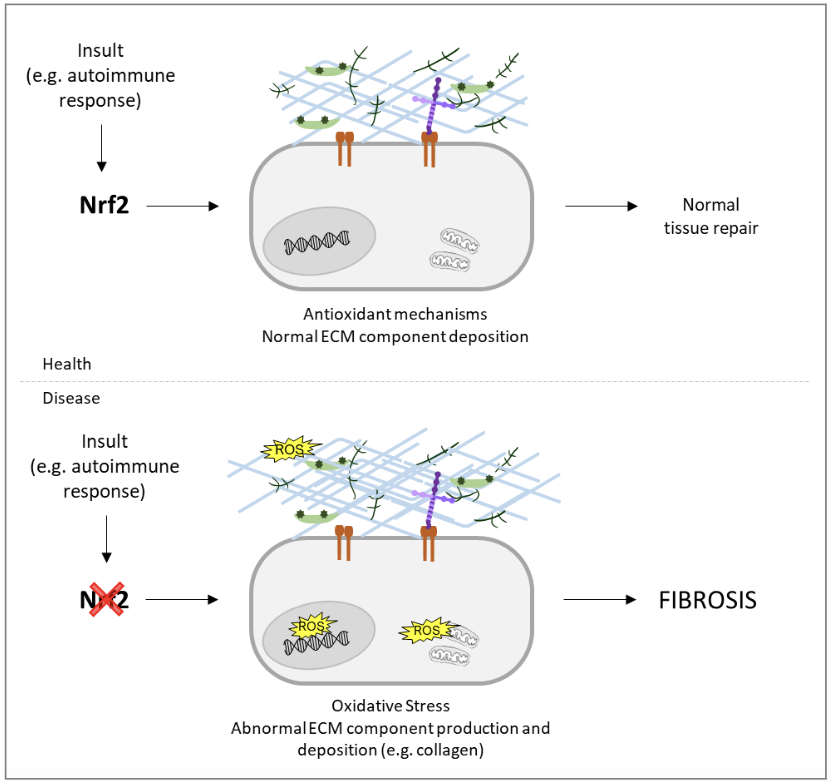
A promising compound is oltipraz, a well-known NRF2 activator, shown to have an impact in preclinical and clinical trials in the reduction of fibrosis. Other NRF2 activators have also been shown to reduce fibrosis in the context of different diseases, including sulforaphane or tanshinone IIA. Being fibrosis such a common outcome in the pathology of a wide range of diseases, research on NFR2 has an enormous potential to uncover new lines of treatment.
Susana G. Martins
WG5 member
Rita Carlos
WG5 leader
University of Lisbon, Portugal
Hot from Pubmed
Nrf2 induces malignant transformation of hepatic progenitor cells by inducing β-catenin expression
The Nrf2 signaling pathway prevents cancer initiation, but genetic mutations that activate this pathway are found in various types of cancer. In this paper, Fragoulis and collaborators identified the Nrf2-β-catenin axis promoting proliferation of hepatic stem cells and triggering tumorigenesis. It was shown that sustained Nrf2 activation induced proliferation and dedifferentiation of a Wnt-responsive perivenular hepatic progenitor cell population, transforming them into metastatic cancer cells with features known from human hepatoblastoma. These results demonstrate that Nrf2-induced upregulation of β-catenin expression and its activation is the underlying mechanism for malignant transformation, representing a breakthrough in the understanding of the role of Nrf2 in regeneration and liver carcinogenesis.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36209041/
UCMSCs-derived exosomal circHIPK3 promotes ulcer wound angiogenesis of diabetes mellitus via miR-20b-5p/Nrf2/VEGFA axis
Experiments confirmed that circular RNAs contributed to the pathogenesis of diabetic foot ulcers (DFUs). Circular RNA homeodomain- interacting protein kinase three (CircHIPK3) was upregulated in type 2 diabetes mellitus (T2DM), but its role in DFU remained unknown. This study by Liang and collaborators demonstrated that high glucose conditions decreased circHIPK3 in HUVECs. Remarkably, umbilical cord mesenchymal stem cell (MSC)-derived exosomal circHIPK3 promoted cell proliferation, migration, and angiogenesis in high glucose-treated HUVECs via downregulation of the miR-20b-5p to upregulate Nrf2 and VEGFA. This modulation resulted in an acceleration of the recovery of diabetic foot ulcers in mice. Therefore, this work showed that exosomal circHIPK3 might be a therapeutic candidate to treat DFU.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36209373/
Collagenolysis-dependent DDR1 signalling dictates pancreatic cancer outcome
Pancreatic ductal adenocarcinoma (PDAC) is a highly desmoplastic, aggressive cancer that frequently progresses and spreads by metastasis to the liver. Cancer-associated fibroblasts, the extracellular matrix and type I collagen (Col I) support or restrain the progression of PDAC and may impede blood supply and nutrient availability. However, the mechanisms remain poorly understood. In this paper, it was shown that matrix-metalloprotease-cleaved Col I (cCol I) and intact Col I (iCol I) exerted opposing effects on PDAC bioenergetics, macropinocytosis, tumour growth and metastasis. Whereas cCol I activated discoidin domain receptor 1 (DDR1)-NF-κB-p62-NRF2 signalling to promote the growth of PDAC, iCol I triggers the degradation of DDR1 and restrains the growth of PDAC. Accordingly, patients whose tumours were enriched for iCol I and expressed low levels of DDR1 and NRF2 had improved median survival compared to those whose tumours have high levels of cCol I, DDR1 and NRF2. The Col I-DDR1-NF-κB-NRF2 mitochondrial biogenesis pathway mediate tumour growth and metastasis and targeting components of this pathway could provide therapeutic opportunities.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36198801/
Lycopene attenuates the inflammation and apoptosis in aristolochic acid nephropathy by targeting the Nrf2 antioxidant system
Lycopene (LYC) is a carotenoid and has antioxidant properties. This study investigated whether LYC attenuates aristolochic acids (AAs)-induced chronic kidney disease in a mice model. AAI induces oxidative stress injury and inhibition of the Nrf2/HO-1 antioxidant signaling pathway in the kidney, which ultimately leads to inflammation and tubular epithelial cell apoptosis. After LYC administration, it activated Nrf2 nuclear translocation and its downstream HO-1 and NQO1 antioxidant signaling pathways. LYC inhibited ROS production by renal tubular epithelial cells and alleviated mitochondrial damage. LYC further modulated the TNF-α/NF-κB signaling cascade, reducing the accumulation of inflammatory factors in the renal interstitium. Moreover, LYC was able to up-regulate the expression of Bcl-2, down-regulate the expression of Bax and inhibit the activation of cleaved forms of Caspase-9 and Caspase-3, attenuating apoptosis. It was concluded that LYC was able to induce the Nrf2 antioxidant signaling pathway to maintain the homeostasis of renal oxidative stress and ultimately attenuate renal inflammatory response and apoptosis. These results suggest that lycopene can be used as a drug to relieve aristolochic acid nephropathy.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36198206/
The molecular activity of cannabidiol in the regulation of Nrf2 system interacting with NF-κB pathway under oxidative stress
Cannabidiol (CBD), the major non-psychoactive phytocannabinoid of Cannabis sativa L., is one of the most studied compounds in pharmacotherapeutic approaches to treat oxidative stress-related disorders such as cardiovascular, metabolic, neurodegenerative, and neoplastic diseases. There have been reports indicating both antioxidant and pro-oxidative effects of CBD. Thus, the mechanism of action of this natural compound in the regulation of Nrf2, which plays the role of the main cytoprotective regulator of redox balance and inflammation under oxidative stress conditions, seems to be particularly important. Accordingly, this paper reviews the CBD-mediated pathways of regulation of the Nrf2 system.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36198205/
Knockout of NRF2 triggers prostate cancer cells death through ROS modulation and sensitizes to cisplatin
Prostate cancer (PCa) represents the second most common cancer in men and affects millions worldwide. Chemotherapy is a common treatment for PCa but the development of resistance is often a problem during therapy. NRF2, besides being the major regulator of antioxidant enzymes, is also involved with drug efflux and detoxification and cancer cells submitted to chemotherapy often promote NRF2 activation to benefit themselves with the cytoprotective response. In this work, it was found that NRF2 knockout in a PCa cell line (DU145) led to a lower IC50 value for cisplatin, suggesting that the knockout sensitized the cells to the treatment. The data presented here support that NRF2 is a mediator of oncogenesis and could be a potential target to sensitize PCa cells to chemotherapy, reinforcing the importance of knowing the specific genetic and biochemical characteristics of the cancer cells for a more effective approach against cancer.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36191155/
FKBP11 rewires UPR signaling to promote glucose homeostasis in type 2 diabetes and obesity
Chronic endoplasmic reticulum (ER) stress and sustained activation of unfolded protein response (UPR) signaling contribute to the development of type 2 diabetes in obesity. UPR signaling is a complex signaling pathway, which is still being explored in many different cellular processes. In this work, it was demonstrated that FK506-binding protein 11 (FKBP11) is severely reduced in the livers of obese mice. Restoring hepatic FKBP11 expression initiated an atypical UPR signaling pathway marked by rewiring of PERK signaling toward NRF2 activation. This alteration in UPR signaling established glucose homeostasis without changing hepatic ER stress, food consumption, or body weight. Therefore, ER stress during obesity could be beneficially rewired to promote glucose homeostasis. These findings may uncover possible new avenues in the development of novel approaches to treat diseases marked by ER stress.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/35793654/
NRF2 mediates melanoma addiction to GCDH by modulating apoptotic signalling
Tumour dependency on specific metabolic signals has been demonstrated and often guided numerous therapeutic approaches. Herein, the authors describe the identification of melanoma addiction to the mitochondrial protein glutaryl-CoA dehydrogenase (GCDH), which functions in lysine metabolism and controls protein glutarylation. GCDH knockdown induced cell death programmes in melanoma cells, an activity blocked by inhibition of the upstream lysine catabolism enzyme DHTKD1. The transcription factor NRF2 mediates GCDH-dependent melanoma cell death programmes. Mechanistically, GCDH knockdown induces NRF2 glutarylation, increasing its stability and DNA binding activity, with a concomitant transcriptional upregulation of ATF4, ATF3, DDIT3 and CHAC1, resulting in cell death. In vivo, inducible inactivation of GCDH effectively inhibited melanoma tumour growth. Correspondingly, reduced GCDH expression correlated with improved survival of patients with melanoma. These findings identify melanoma cell addiction to GCDH, limiting apoptotic signalling by controlling NRF2 glutarylation. Inhibiting the GCDH pathway could thus represent a therapeutic approach to treat melanoma.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36050469/
Maladaptive positive feedback production of ChREBPβ underlies glucotoxic β-cell failure
Preservation and expansion of β-cell mass is a therapeutic goal for diabetes. In this paper, Katz and colleagues demonstrated that transient positive feedback induction of carbohydrate response-element binding protein (ChREBPβ) is necessary for adaptive β-cell expansion in response to metabolic challenges. Conversely, chronic excessive β-cell-specific overexpression of ChREBPβ results in loss of β-cell identity, apoptosis, loss of β-cell mass, and diabetes. Importantly, ChREBPβ-mediated cell death is mitigated by overexpression of the alternate CHREBP gene product, ChREBPα, or by activation of the antioxidant Nrf2 pathway.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/35908073/
An open-label pilot study of recombinant granulocyte-colony stimulating factor in Friedreich’s ataxia
Friedreich’s ataxia (FA) is an inherited progressive neurodegenerative disease for which there is no proven disease-modifying treatment. In this study the authors performed an open-label, pilot study of recombinant human granulocyte-colony stimulating factor (G-CSF) administration in seven people with FA (EudraCT: 2017-003084-34); each participant receiving a single course of G-CSF (Lenograstim; 1.28 million units per kg per day for 5 days). The primary outcome is peripheral blood mononuclear cell frataxin levels over a 19-day period. The secondary outcomes include safety, haematopoietic stem cell (HSC) mobilisation, antioxidant levels and mitochondrial enzyme activity. The trial meets pre-specified endpoints. We show that administration of G-CSF to people with FA is safe. Mobilisation of HSCs in response to G-CSF is comparable to that of healthy individuals. Notably, sustained increases in cellular frataxin concentrations and raised PGC-1α and Nrf2 expression are detected. These findings show potential for G-CSF therapy to have a clinical impact in people with FA.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/35945193/
Joana Miranda
University of Lisbon
Portugal