
NRF2 in COST Action 20121
Our quarterly newsletter attempts to provide our latest news and also aims at becoming a forum for analysis of relevant topics on the field of NRF2, and provide comments to some of the most relevant articles published during the quarter. Previous newsletters can be accessed at:
https://benbedphar.org/our-first-newsletter/
https://benbedphar.org/issue-2-abril-2022/
https://benbedphar.org/issue-3-july-2022/
https://benbedphar.org/issue-4-october-2022/
https://benbedphar.org/issue-5-january-2023/
On April 19th we had our Management Committee (April 19th) and Scientific meeting (April 20-21) that was held at Zagreb (Croatia) and organized by Prof. Ana Cipak and her team. We discussed the progress of the Action and planed new activities towards the end of this second grant period. During the scientific meeting we had 32 excellent communications. The programme can be found at:
https://benbedphar.org/4th-benbedphar-meeting-from-physiology-to-pathology/
One of the most successful BenBedPhar activities is being the funding of Short-Term Scientific Missions. This COST tool is allowing the interaction among many different EU laboratories by providing short term visits of young researchers to collaborative laboratories. We hope that these collaborative efforts soon will results in new experimental work towards the understanding of the role of NRF2 in physiology and pathology.
The Annual meeting of the SFRR-Europe will be held in Vienna during June 6-9. Several BenBedPhar members supported the nomination of Prof. Masayuki Yamamoto for the Annual Award Lecture of the society, and we are very honoured to see that he will receive this award on June 8th.
The 21st Biennial Congress of the SFRR-International will take place in Punta del Este, Uruguay on November 15-18. Our application for a symposium on NRF2 has been approved. The chair of the symposium will be Prof. Giovanni Mann (UK member of our Management Committee) and Prof. Henry J. Forman. The speakers will be Donna Zhang (BenBedPhar member from USA) and myself (BenBedPhar Chair). We thank the Society for this opportunity to disseminate our work in the Action.
Another excellent news is that our Grant Holder Manager and Gender Equality Officer, Dr. Elana Milanesi, received the L’Oreal award for year 2022 in Romania. Congratulations Elena! See details at: https://www.hackathons.ro/loreal-romania-anunta-castigatoarele-burselor-private-loreal-unesco-pentru-femeile-din-stiinta/
Antonio Cuadrado
Chair of COST Action 20121, BenBedPhar
Autonomous University of Madrid
Comments from the Working Groups
NRF2 in Regulation of Iron Metabolism: Focus on Cardiovascular Disorders and Cardioprotection
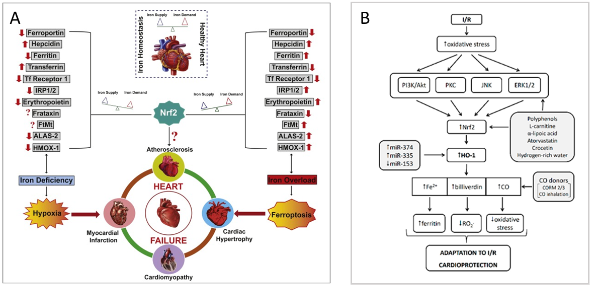
Disorders of iron metabolism are involved in the pathological mechanisms of noncommunicable diseases, such as type 2 diabetes, obesity, non-alcoholic fatty liver disease, cardiovascular disorders and others. Dysregulation of iron metabolism can result in both low and high iron levels – both of them are potentially dangerous to the tissues, especially to the heart (1). In general, iron metabolism in mammals is tightly regulated on the systemic level by the hepatic hormone hepcidin (HEP), which is the central regulatory molecule of systemic iron homeostasis. HEP is released also in the heart, it is assumed that it is involved in the local regulation of iron metabolism. On a cellular level, iron metabolism is regulated via the hypoxia-inducible factor 1 (HIF1) and iron regulatory proteins 1 and 2 (IRP1/2). IRP1 function (either as IRP or as aconitase 2) is further affected by oxidative stress, nitric oxide and hydrogen sulfide (2), all of which can modify the intracellular concentration of iron. Iron-induced redox imbalance is present in various diseases, since free iron readily transforms between Fe2+ and Fe3+ via the Fenton reaction, leading to reactive oxygen species production. In addition to the activation of NRF2-mediated antioxidant and cytoprotective mechanisms, the involvement of NRF2 in the regulation of several genes related to heme synthesis and catabolism, iron storage, transport and export, including ferrochelatase (FECH) aminolevulinic acid synthase (ALAS), heme oxygenase 1 (HMOX1), transferrin receptor 1 (TFR1), ferritin heavy chain (FTH1), ferritin light chain (FTL) and ferroportin (FPN1) has been described (1, 3, 4). NRF2 also controls HEP synthesis via ARE segment in HEP gene (HAMP) promoter and indirectly via NRF2-induced bone morphogenetic protein 6 (BMP6) synthesis. Moreover, NRF2 also regulates the sensitivity of cells to iron-dependent cell death ferroptosis via the regulation of the GPX family of proteins, namely GPX4. Iron is essential in normal cardiac function and recent investigations have demonstrated that iron imbalance, iron-dependent oxidative stress and ferroptosis are common in conditions of heart failure (HF), ischemia/reperfusion (I/R) injury as well as myocardial infarction, coronary artery angioplasty or heart transplantation (1, 3). Data suggest that the HEP/FPN/NFR2 axis may act as an “epicenter” linking iron metabolism to redox changes (3) while targeting NRF2-mediated antioxidant signaling is rational to combat iron-mediated oxidative stress in conditions of HF (Figure 1A) or I/R injury (Figure 1B). In the context of cardioprotection, a variety of natural and synthetic substances have been shown to exert their cardioprotective effects via NRF2/HO-1 activation resulting in the modulation of cellular iron metabolism. Thus, with any pharmacological modulation of NRF2 production, whether with drugs or nutritional supplements, it is necessary to keep in mind the possible alterations in iron metabolism and the potential effects on heart function.
References:
1. Ravingerová T, Kindernay L, Barteková M, Ferko M, Adameová A, Zohdi V, Bernátová I, Ferenczyová K, Lazou A. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. https://doi.org/10.3390/ijms21217889
2. Lushchak OV, Piroddi M, Galli F, Lushchak VI. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014; 19(1): 8–15. Doi: 10.1179/1351000213Y.0000000073
3. Jayakumar D, S Narasimhan KK, Periandavan K. Triad role of hepcidin, ferroportin, and Nrf2 in cardiac iron metabolism: From health to disease. J. Trace Elem. Med. Biol. 2022 Jan;69:126882. https://doi.org/10.1016/j.jtemb.2021.126882
4. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox. Signal. 2018 Dec 10; 29(17): 1756–1773. doi: 10.1089/ars.2017.7176
Iveta Bernatova
WG1 member
Centre of Experimental Medicine, Slovak Academy of Sciences, Bratislava, Slovakia,
A potent and selective KEAP1-NRF2 protein-protein interaction inhibitor with hepatic anti-fibrotic activity in mice
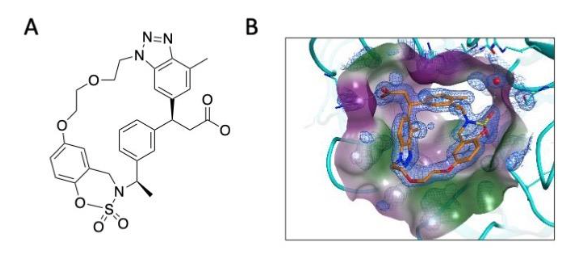
With the aim to increase the selectivity for KEAP1 and reduce the cytotoxicity associated with high concentrations of electrophiles, a number of non-electrophilic NRF2 activators have been developed. These compounds are designed to bind to the Kelch domain of KEAP1 and conform to the Hinge-Latch model of NRF2 regulation by KEAP1, whereby the DLGex-binding motif of NRF2 dissociates from KEAP1 as a latch, while the ETGE motif remains attached to KEAP1 as a hinge (Horie et al. 2021). Many KEAP1-NRF2 protein-protein interaction (PPI) inhibitors have high affinities for KEAP1-Kelch in vitro, with some showing potencies similar to the classical NRF2 activator, the electrophile sulforaphane in cell-based assays (Dayalan Naidu et al. 2021). However, the in vivo bioavailability and efficacy of most of these compounds following oral administration is limited by unfavourable absorption, distribution, metabolism, excretion and pharmacokinetics (ADME/PK). One such compound is KI-696, which due to its low oral bioavailability, requires intravenous infusion (Davies et al. 2016). Using KI-696 compound as a starting point, Seedorf et al (2022) replaced the benzoxathiazepine and benzoyl scaffolds with benzoxathiazine. They hypothesized that position 6 of the benzene ring of benzoxathiazine would allow linking the aromatic ring with the N-alkyl group of benztriazole and would make possible various substitution patterns on the linker. This approach led to the design of S217879 (Figure 2), which binds to the Kelch domain of KEAP1 with high affinity (Kd = 4.15 nM) and selectivity and activates NRF2 in cells at nanomolar concentrations, as well as in the mouse liver in vivo. In mice on methionine and choline-deficient diet, a model of non-alcoholic steatohepatitis (NASH), orally-administered S217879 for two weeks resulted in a dose-dependent reduction in disease activity score. Moreover, this compound was also effective in established disease in a diet-induced obesity NASH mouse model, where (30 mg/kg, p.o., q.d.) reduced both disease activity score and liver fibrosis. These results suggest that non-electrophilic KEAP1-NRF2 PPI inhibitors can be developed for the treatment of liver disease.
References
Horie et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun Biol. 2021; 4: 576.
Davies et al. Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J Med Chem. 2016; 59: 3991-4006.
Dayalan Naidu et al. The isoquinoline PRL-295 increases the thermostability of Keap1 and disrupts its interaction with Nrf2. iScience. 2021; 25: 103703.
Seedorf et al. Selective disruption of NRF2-KEAP1 interaction leads to NASH resolution and reduction of liver fibrosis in mice. JHEP Rep. 2022; 5: 100651.
Prof. Albena Dinkova-Kostova
WG2 leader
University of Dundee, UK
The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis
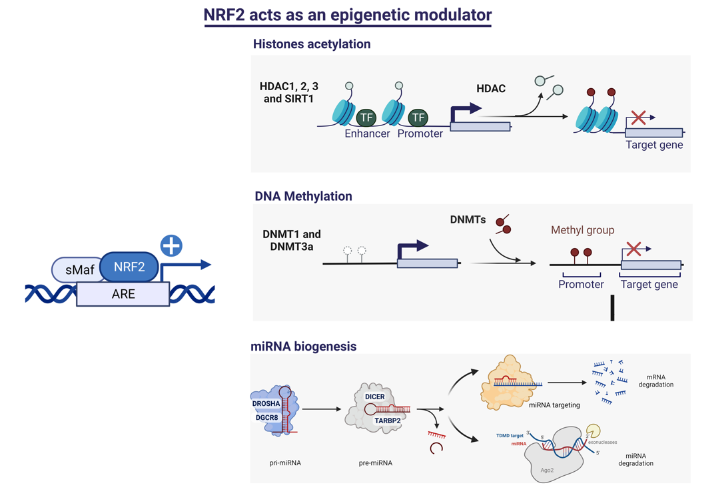
The epigenetic regulation of gene expression is a complex and tightly regulated process that defines cellular identity and is associated with health and disease processes. Oxidative stress is capable of inducing epigenetic modifications. The transcription factor NRF2 (nuclear factor erythroid-derived 2-like 2) is a master regulator of cellular homeostasis, regulating genes bearing antioxidant response elements (AREs) in their promoters. Here, we report the identification of ARE sequences in the promoter regions of genes encoding several epigenetic regulatory factors, such as histone deacetylases (HDACs), DNA methyltransferases (DNMTs), and proteins involved in microRNA biogenesis. In this research, we study this possibility by integrating bioinformatic, genetic, pharmacological, and molecular approaches (Figure 3). We found ARE sequences in the promoter regions of genes encoding several HDACs, DNMTs, and proteins involved in miRNA biogenesis. We confirmed that NRF2 regulates the production of these genes by studying NRF2-deficient cells and cells treated with dimethyl fumarate (DMF), an inducer of the NRF2 signaling pathway. In addition, we found that NRF2 could be involved in the target RNA-dependent microRNA degradation (TDMD) of miR-155-5p through its interaction with Nfe2l2 mRNA. Our data indicate that NRF2 has an epigenetic regulatory function, complementing its traditional function and expanding the regulatory dimensions that should be considered when developing NRF2-centered therapeutic strategies.
References
Silva-Llanes I, Shin CH, Jiménez-Villegas J, Gorospe M, Lastres-Becker I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants (Basel). 2023 Mar 4;12(3):641. doi: 10.3390/antiox12030641.
Isabel Lastres Becker
WG3 member
Instituto de Investigaciones Biomedicas Alberto Sols, CISC-UAM, Madrid, Spain
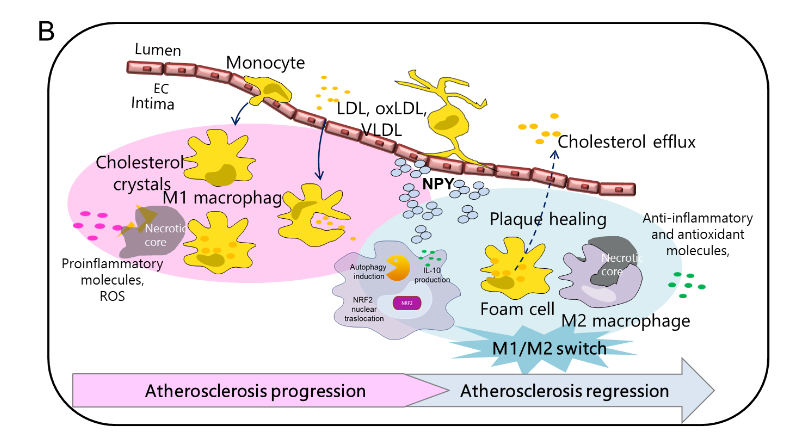
Neuropeptide Y promotes human M2 macrophage polarization and enhances p62/SQSTM1-dependent autophagy and NRF2 activation
Neuropeptide Y (NPY) is an abundantly expressed peptide capable of modulating innate and adaptive immune responses and regulating chemotaxis and cytokine secretion by macrophages. Abnormal regulation of NPY is involved in the development of atherosclerosis. The inflammatory infiltrate within atherosclerotic plaque is characterized by accumulation of macrophages, which are subject to reprogram their phenotypes in response to environmental signals. Macrophage number and phenotype influence plaque fate. Here, we investigated the effect of NPY on the changes in phenotype and functions of human macrophages, from the pro-inflammatory phenotype M1 to the reparative M2, indicative of atherosclerosis regression or stabilization. Human monocytes were differentiated in vitro into macrophages with M-CSF (M0) and polarized towards an M1 phenotype with IFN-γ plus LPS M(IFN-γ/LPS) or M2 with IL-10 (M IL-10) and further challenged with NPY (10−7–10−9 M) for 8–36 h. Cell phenotype and functions were analyzed by immunofluorescence and immunochemical analyses. NPY affected macrophage surface markers and secretome profile expression, thus shifting macrophages toward an M2-like phenotype. NPY also prevented the impairment of endocytosis triggered by the oxysterol 7-keto-cholesterol (7KC) and prevented 7KC-induced foam cell formation by reducing the lipid droplet accumulation in M0 macrophages. NPY-treated M0 macrophages enhanced the autophagosome formation by upregulating the cell content of the autophagy markers LC3-II and p62-SQSTM1, increased activation of the anti-oxidative transcription factor NRF2 (NF-E2-related factor 2), and subsequently induced its target gene HMOX1 that encodes heme oxygenase-1 (Figure 4A). Our findings indicate that NPY has a cytoprotective effect with respect to the progression of the inflammatory pathway, both enhancing p62/SQSTM1-dependent autophagy and the NRF2–antioxidant signalling pathway in macrophages. NPY signalling may have a crucial role in tissue homeostasis in host inflammatory responses through the regulation of macrophage balance and functions within atherosclerosis (Figure 4B).
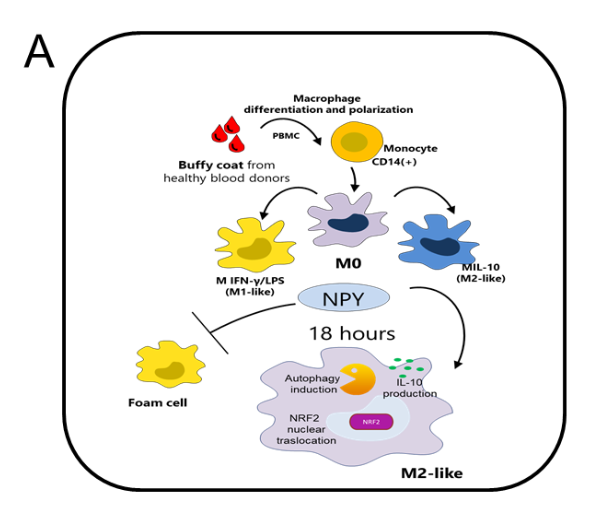
Figure 4. NPY is able to tune M1/M2
macrophage balance towards a M2-like
phenotype. The study provides a critical link
between autophagy dysregulation and
prolonged NRF2 signaling in a p62-dependent
manner and explains, at least in part, the close
relationship of NPY and macrophages with the
pathophysiological mechanisms underlying
arteriosclerotic cardiovascular disease [1], as
well as the contradictory results obtained in
animal models of atherosclerosis deficient for
NPY [2] or NRF2 [3]. However, further studies
are required to dissect the effects of NPY on
p62-dependent NRF2 activation.
References
1. Zhu, P.; Sun, W.; Zhang, C.; Song, Z.; Lin, S. The role of neuropeptide Y in the pathophysiology of atherosclerotic cardio-vascular disease. Int. J. Cardiol. 2016, 220, 235–241.
2. Qin, Y.Y.; Huang, X.R.; Zhang, J.; Wu, W.; Chen, J.; Wan, S.; Yu, X.Y.; Lan, H.Y. Neuropeptide Y attenuates cardiac remodeling and deterioration of function following myocardial infarction. Mol. Ther. 2022, 30, 881–897.
3. Ruotsalainen, A.K.; Lappalainen, J.P.; Heiskanen, E.; Merentie, M.; Sihvola, V.; Näpänkangas, J.; Lottonen-Raikaslehto, L.; Kansanen, E.; Adinolfi, S.; Kaarniranta, K.; et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc. Res. 2019, 115, 243–254.
Brigitta Buttari
WG5 Leader, on behalf of authors
Hot from Pubmed
Role of HNF4alpha-cMyc interaction in liver regeneration and recovery after acetaminophen-induced acute liver injury
Overdose of acetaminophen (APAP) is the major cause of acute liver failure in the Western world. Herein, it is reported a novel signaling interaction between Hepatocyte Nuclear Factor 4 alpha (HNF4α) cMyc and Nrf2 during liver injury and regeneration after APAP overdose. APAP-induced liver injury and regeneration were studied in male C57BL/6J (WT) mice, hepatocyte specific HNF4α knockout mice (HNF4α -KO) and HNF4α-cMyc double knockout mice (DKO). C57BL/6J mice treated with 300 mg/kg maintained nuclear HNF4α expression and exhibited liver regeneration, resulting in recovery. However, treatment with 600 mg/kg APAP, where liver regeneration was inhibited and recovery was delayed, showed rapid decline in HNF4α expression. HNF4α-KO mice developed significantly higher liver injury due to delayed GSH recovery after APAP overdose. HNF4α-KO mice also exhibited significant induction of cMyc, and deletion of cMyc in HNF4α-KO mice (DKO mice) reduced APAP-induced liver injury. The DKO mice had significantly faster GSH replenishment due to rapid induction in Gclc and Gclm genes. Co-IP and ChIP analysis revealed that HNF4α interacts with Nrf2 and affects its DNA binding. Further, DKO mice showed significantly faster initiation of cell proliferation resulting in rapid liver regeneration and recovery. These data show that HNF4α interacts with Nrf2 and promotes GSH replenishment aiding in recovery from APAP-induced liver injury, a process inhibited by cMyc. These studies indicate that maintaining HNF4α function is critical for regeneration and recovery after APAP overdose.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37021787/
ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis
Hepatocellular carcinoma (HCC), a leading cause of cancer death, is associated with viral hepatitis, non-alcoholic and alcoholic steatohepatitis (NASH, ASH), all of which trigger endoplasmic reticulum (ER) stress, hepatocyte death, inflammation, and compensatory proliferation. Using ER stress-prone MUP-uPA mice, we established that ER stress and hypernutrition cooperate to cause NASH and HCC, but the contribution of individual stress effectors, such as ATF4, to HCC and their underlying mechanisms of action remained unknown. Hepatocyte-specific ATF4 deficient MUP-uPA mice (MUP-uPA/Atf4Δhep) and control MUP-uPA/Atf4F/F mice were fed high fat diet (HFD) to induce NASH-induced HCC, and Atf4F/F and Atf4Δhep mice were injected with diethylnitrosamine (DEN) to model carcinogen-induced HCC. Histological, biochemical, and RNA sequencing analyses were performed to identify and define the role of ATF4-induced SLC7A11 expression in hepatocarcinogenesis. Reconstitution of SLC7A11 in ATF4-deficient primary hepatocytes and mouse livers was used to study its effects on ferroptosis and HCC development. Hepatocyte ATF4 ablation inhibited hepatosteatosis, but increased susceptibility to ferroptosis, resulting in accelerated HCC development. Although ATF4 activates numerous genes, ferroptosis susceptibility and hepatocarcinogenesis were reversed by ectopic expression of a single ATF4 target, Slc7a11, coding for a subunit of the cystine-glutamate antiporter xCT, which is needed for glutathione (GSH) synthesis. A ferroptosis inhibitor also reduced liver damage and inflammation. ATF4 and SLC7A11 amounts were positively correlated in human HCC and livers of NASH patients. Despite ATF4 being upregulated in established HCC, it serves an important protective function in normal hepatocytes. By maintaining glutathione production ATF4 inhibits ferroptosis-dependent inflammatory cell death, which is known to promote compensatory proliferation and hepatocarcinogenesis. Ferroptosis inhibitors or ATF4 activators may also blunt HCC onset.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36996941/
NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer
Multiple cancers regulate oxidative stress by activating the transcription factor NRF2 through mutation of its negative regulator, KEAP1. NRF2 has been studied extensively in KEAP1-mutant cancers; however, the role of this pathway in cancers with wild-type KEAP1 remains poorly understood. To answer this question, we induced NRF2 via pharmacological inactivation of KEAP1 in a panel of 50+ non-small cell lung cancer cell lines. Unexpectedly, marked decreases in viability were observed in >13% of the cell lines-an effect that was rescued by NRF2 ablation. Genome-wide and targeted CRISPR screens revealed that NRF2 induces NADH-reductive stress, through the upregulation of the NAD+-consuming enzyme ALDH3A1. Leveraging these findings, we show that cells treated with KEAP1 inhibitors or those with endogenous KEAP1 mutations are selectively vulnerable to Complex I inhibition, which impairs NADH oxidation capacity and potentiates reductive stress. Thus, we identify reductive stress as a metabolic vulnerability in NRF2-activated lung cancers.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36841242/
ceAF Ameliorates Diabetic Wound Healing by Alleviating Inflammation and Oxidative Stress via TLR4/NF-κB and Nrf2 Pathways.
With the rise in diabetes incidence, diabetic foot ulcers have become the most common clinically chronic refractory wounds. Persistent chronic inflammation is a typical feature of diabetic cutaneous wounds, and diabetic wound healing can be improved by alleviating inflammation and oxidative stress. Chick early amniotic fluids (ceAF) consist of native conglutinant substances with balanced amounts of growth factors, cytokines, and chemokines. However, whether ceAF modulates inflammation and oxidative stress and thus promotes diabetic wound healing remains unknown. Herein, the authors aimed to explore the effect of ceAF on wound healing and its molecular mechanism. Topical administration of ceAF improved M2 macrophage polarization and inflammatory response in the wound tissues, thereby ameliorating delayed wound healing. Histological improvement could be observed in the grade of inflammation, collagen deposition, and neovascularization in wound edge tissues. ceAF also increased M2 macrophage-specific markers expression and exogenous ceAF suppressed LPS-induced cellular inflammatory response in vitrohigh glucose environment. Additionally, ceAF could activate TLR4/NF-κB and Nrf2 signal transductions to promote M2 macrophage. In summary, ceAF downregulates inflammatory response, regulates M2 macrophage transition via TLR4/NF-κB and Nrf2 signaling pathways, and thus improves diabetic wound healing.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37064758/
Optimal combination of arsenic trioxide and copper ions to prevent autoimmunity in a murine HOCl-induced model of systemic sclerosis.
Systemic sclerosis (SSc) is a rare chronic autoimmune disease characterized by diffuse fibrosis of the skin and internal organs and vascular abnormalities. The etiology and physiopathology are complex due to the heterogeneity of its overall clinical presentation. Arsenic trioxide (ATO) has been proven to be effective against SSc, sclerodermatous Graft-versus-Host Disease, multiple sclerosis, Crohn’s disease or systemic lupus erythematosus animal models and has demonstrated promising effects in human clinical trials. Its efficacy was shown to be related at least in part to the generation of Reactive Oxygen Species (ROS) and the selective deletion of activated immune cells and fibroblasts. However, ATO can induce some adverse effects that must be considered, especially when used for the treatment of a chronic disease. The authors evaluated, in vitro and in a mouse model of SSc, the improved efficacy of ATO when associated with a Fenton-like divalent cation, namely copper chloride (CuCl2), also known to trigger the production of ROS. In preliminary experiments in vitro, ATO 1 µM + CuCl2 0.5 µM increased ROS production and increased apoptosis of NIH 3T3 murine fibroblasts compared to 1 µM ATO alone. In vivo, in the HOCl-induced mouse model of SSc, co-treatment with ATO 2.5 μg/g + CuCl2 0.5 μg/g significantly alleviated clinical signs such as the thickening of the skin (p<0.01) and cutaneous fibrosis, in a manner equivalent to treatment with ATO 5 µg/g. The results provide evidence that co-treatment with ATO 2.5 μg/g + CuCl2 0.5 μg/g decreases the number of B cells and the activation of CD4+ T lymphocytes. The co-treatment substantially blocks the NRF2 signaling pathway, increases H2O2 production and results in the improvement of the health status of mice with experimental SSc. In conclusion, copper combined with ATO treatment halved the concentration of ATO needed to obtain the same effect as a high dose of ATO alone for the treatment of SSc mice. The strategy of using lower doses of drugs with different mechanisms of action in combination has many potential advantages, the first being to lessen the potential side effects induced by ATO, a drug with side effects quickly increased with dosage.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37063915/
Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload.
Hepatic cholesterol overload promotes steatohepatitis. Insufficient understanding of liver stress defense impedes therapy development. Here, we elucidate the role of stress defense transcription factors, nuclear factor erythroid 2 related factor-1 (NRF1) and -2 (NRF2), in counteracting cholesterol-linked liver stress. Using a diet that increases liver cholesterol storage, expression profiles and phenotypes of liver from mice with hepatocyte deficiency of NRF1, NRF2, or both are compared with controls, and chromatin immunoprecipitation sequencing is undertaken to identify target genes. Results show NRF1 and NRF2 co-regulate genes that eliminate cholesterol and mitigate inflammation and oxidative damage. Combined deficiency, but not deficiency of either alone, results in severe steatohepatitis, hepatic cholesterol overload and crystallization, altered bile acid metabolism, and decreased biliary cholesterol. Moreover, therapeutic effects of NRF2-activating drug bardoxolone require NRF1 and are supplemented by NRF1 overexpression. Thus, we discover complementary gene programming by NRF1 and NRF2 that counteract cholesterol-associated fatty liver disease progression.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37060561/
The integrated stress response is activated in the salivary glands of Sjögren’s syndrome patients
Primary Sjögren’s syndrome (SS) is an autoimmune exocrinopathy that affects the structure and function of salivary and lachrymal glands. Labial salivary gland (LSG) acinar cells from SS patients lose cellular homeostasis and experience endoplasmic reticulum and oxidative stress. The integrated cellular stress response (ISR) is an adaptive pathway essential for restoring homeostasis against various stress-inducing factors, including pro-inflammatory cytokines, and endoplasmic reticulum and oxidative stress. ISR activation leads eIF2α phosphorylation, which transiently blocks protein synthesis while allowing the ATF4 expression, which induces a gene expression program that seeks to optimize cellular recovery. PKR, HRI, GCN2, and PERK are the four sentinel stress kinases that control eIF2α phosphorylation. Dysregulation and chronic activation of ISR signaling have pathologic consequences associated with inflammation. Here, it was analyzed the activation of the ISR in LSGs of SS-patients and non-SS sicca controls, determining the mRNA, protein, and phosphorylated-protein levels of key ISR components, as well as the expression of some of ATF4 targets. Moreover, we performed a qualitative characterization of the distribution of ISR components in LSGs from both groups and evaluated if their levels correlate with clinical parameters. It was observed that the four ISR sensors are expressed in LSGs of both groups. However, only PKR and PERK showed increased expression and/or activation in LSGs from SS-patients. eIF2α and p-eIF2α protein levels significantly increased in SS-patients; meanwhile components of the PP1c complex responsible for eIF2α dephosphorylation decreased. ATF4 mRNA levels were decreased in LSGs from SS-patients along with hypermethylation of the ATF4 promoter. Despite low mRNA levels, SS-patients showed increased levels of ATF4 protein and ATF4-target genes involved in the antioxidant response. The acinar cells of SS-patients showed increased staining intensity for PKR, p-PKR, p-PERK, p-eIF2α, ATF4, xCT, CHOP, and NRF2. Autoantibodies, focus score, and ESSDAI were correlated with p-PERK/PERK ratio and ATF4 protein levels. In summary, the results showed an increased ISR activation in LSGs of SS-patients. The increased protein levels of ATF4 and ATF4-target genes involved in the redox homeostasis could be part of a rescue response against the various stressful conditions to which the LSGs of SS-patients are subjected and promote cell survival.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37035319/
Chronic hyperglycaemia increases the vulnerability of the hippocampus to oxidative damage induced during post-hypoglycaemic hyperglycaemia in a mouse model of chemically induced type 1 diabetes
Chronic hyperglycaemia and recurrent hypoglycaemia are independently associated with accelerated cognitive decline in type 1 diabetes. Recurrent hypoglycaemia in rodent models of chemically induced (streptozotocin [STZ]) diabetes leads to cognitive impairment in memory-related tasks associated with hippocampal oxidative damage. This study examined the hypothesis that post-hypoglycaemic hyperglycaemia in STZ-diabetes exacerbates hippocampal oxidative stress and explored potential contributory mechanisms. Evidence of hippocampal oxidative damage was most marked in mice with STZ-diabetes exposed to post-hypoglycaemic hyperglycaemia; these mice also showed induction of Nrf2 and the Nrf2 transcriptional targets Sod2 and Hmox-1. In this group, hypoglycaemia induced a significant upregulation of proteins involved in alternative fuel provision, reductive biosynthesis and degradation of damaged proteins, and a significant downregulation of proteins mediating the stress response. Key differences emerged between mice with and without STZ-diabetes following recovery from hypoglycaemia in proteins mediating the stress response and reductive biosynthesis. Overall, there is a disruption of the cellular response to a hypoglycaemic challenge in mice with STZ-induced diabetes that is not seen in wild-type non-diabetic animals. The chronic hyperglycaemia of diabetes and post-hypoglycaemic hyperglycaemia act synergistically to induce oxidative stress and damage in the hippocampus, possibly leading to irreversible damage/modification to proteins or synapses between cells. In conclusion, recurrent hypoglycaemia in sub-optimally controlled diabetes may contribute, at least in part, to accelerated cognitive decline through amplifying oxidative damage in key brain regions, such as the hippocampus.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37015997/
Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis
Ferroptosis, an iron-dependent lipid peroxidation-driven programmed cell death, is closely related to cancer therapy. The development of druggable ferroptosis inducers and their rational application in cancer therapy are critical. Here, we identified Tubastatin A, an HDAC6 inhibitor as a novel druggable ferroptosis inducer through large-scale drug screening. Tubastatin A directly bonded to GPX4 and inhibited GPX4 enzymatic activity through biotin-linked Tubastatin A putdown and LC/MS analysis, which is independent of its inhibition of HDAC6. In addition, our results showed that radiotherapy not only activated Nrf2-mediated GPX4 transcription but also inhibited lysosome-mediated GPX4 degradation, subsequently inducing ferroptosis tolerance and radioresistance in cancer cells. Tubastatin A overcame ferroptosis resistance and radioresistance of cancer cells by inhibiting GPX4 enzymatic activity. More importantly, Tubastatin A has excellent bioavailability, as demonstrated by its ability to significantly promote radiotherapy-induced lipid peroxidation and tumour suppression in a mouse xenograft model. Our findings identify a novel druggable ferroptosis inducer, Tubastatin A, which enhances radiotherapy-mediated antitumor effects. This work provides a compelling rationale for the clinical evaluation of Tubastatin A, especially in combination with radiotherapy.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36989572/
Toxicity manifestations and sex differences due to MARTA olanzapine
Olanzapine is widely used as a treatment for schizophrenia and other psychiatric disorders. Its metabolic side effects, including weight gain and hyperglycemia, are a clinical problem; however, their full mechanism is not yet clearly understood. Recently, it was reported that the accumulation of oxidative stress in the hypothalamus may cause obesity and diabetes mellitus. Epidemiologically, metabolic side effects are known to be more likely to occur in women. In the present study, the authors investigated and tested the hypothesis that olanzapine induces oxidative stress in the hypothalamus and induces metabolic side effects. It was also examined its association with sex differences. Olanzapine was administered intraperitoneally to male and female C57BL/6 mice, and the expression levels of oxidative stress-responsible genes in the hypothalamus and cerebral cortex were measured by qRT-PCR. In addition, olanzapine was administered intraperitoneally to C57BL/6 and Nrf2 KO mice, and the expression level of total glutathione was measured. Gene expressions induced by the Keap1-Nrf2-regulated system showed different responses to olanzapine for each gene. Under the conditions of this experiment, cystine-glutamate transporter was decreased although heme oxygenase-1 and γ-glutamylcysteine synthetase were increased. It was also clear that these responses were not hypothalamus-specific. Long-term feeding with olanzapine suppressed weight gain in males but not females. No glucose intolerance was observed at 13 weeks of administration. Furthermore, deaths occurred only in females. In conclusion, this study failed to provide evidence that olanzapine induces oxidative stress in a hypothalamic-specific manner. Instead, sex differences were observed in response to long-term and high-dose olanzapine administration, suggesting that individual susceptibility to olanzapine toxicity occurred in female mice.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/37005277/
Vaping Induced Cannabidiol (CBD) Oxidation Product CBD Quinone Forms Protein Adducts with KEAP1 and Activates KEAP1-Nrf2 Genes
Cannabidiol (CBD) vaping products have become widely available in the U.S. since their legalization in 2018. However, little is known about their respiratory health effects. Here we show that aerosolization of commercial CBD vaping products generates a reactive CBD quinone (CBDQ) which forms adducts with protein cysteine residues. Using click chemistry and a novel in vitro vaping product exposure system (VaPES), we further demonstrate that CBDQ forms adducts with human bronchial epithelial cell proteins including Keap1 and activates KEAP1-Nrf2 stress response pathway genes. These results suggest that vaping CBD alters protein function and induces cellular stress pathways in the lung.
Access to the original article: https://pubmed.ncbi.nlm.nih.gov/36999736/
Grants
Michelson Prizes: Next Generation Grants – immunology and vaccine development; deadline for submission June 11th, 2023
The Lundbeck Foundation Young Investigator Prize – Deadline June 29th, 2023 https://lundbeckfonden.com/uddelinger-priser/ansoegere/apply-grants/young-investigator-prize
Drug development RFP – Alzheimer’s disease; Upcoming deadline for submission: May 19, 2023 (Letter of Intent), July 28, 2023 (Invited Full Proposal). https://www.alzdiscovery.org/research-and-grants/funding-opportunities/drug-development-rfp
CurePSP – Neurodegenerative diseases; Deadline for submission is October, 2023 (date to be anounced). https://www.psp.org/iwanttolearn/grants/#page-section-research-grants
HORIZON-HLTH-2023-DISEASE-07-01– Deadline for submission September 19th, 2023 https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/horizon-hlth-2023-disease-07-01
HORIZON-HLTH-2024-DISEASE-03-13-two-stage Deadline for submission September 19th, 2023 https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/horizon-hlth-2024-disease-03-08-two-stage
HORIZON-HLTH-2024-TOOL-05-06-two-stage – Deadline for submission September 19th, 2023 https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/horizon-hlth-2024-tool-05-06-two-stage
HORIZON-HLTH-2024-DISEASE-03-13-two-stage – Deadline for submission September 19th, 2023 https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/horizon-hlth-2024-disease-03-13-two-stage
European Commission funding opportunities: https://research-and-innovation.ec.europa.eu/funding_en
European Association for Cancer Research (EACR) Travel Fellowships: https://www.eacr.org/travel-fellowships
Pfizer funding opportunities: https://www.pfizer.com/about/programs-policies/grants/competitive-grants
Networks
EACR Find a collaboration: https://www.eacr.org/content/online-collaborator.php
EIT Health: https://eithealth.eu/
Klaster LifeScience Krakow Foundation: https://lifescience.pl